Cheminformatics Tutorials - Herong's Tutorial Examples - v2.03, by Herong Yang
Wedge-Hash Bond Changed by Open Babel
This section provides a tutorial example demonstrating how Open Babel 'correcting' wedge-hash bond representations of stereo centers from molecule input data.
Sometimes, you may have an Wedge-Hash bond in the input data to specify a stereo center. When you convert the input data with Open Babel, you will see that the stereo center is identified with a different Wedge-Hash bond.
For example, the following SDF/Mol file, L-Alanin-2D.sdf, specifies atom #2 as a stereo center with a wedge bond, #1, connecting the stereo center to atom "N".
L-Alanin
HerongYang.com
2D Stereoinformation: Stereo Code
6 5 0 0 1 0 0 0 0 0999 V2000
3.6373 2.1000 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
2.4249 1.4000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4249 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2124 2.1000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2124 3.5000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 1.4000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2 1 1 1 0 0 0
3 2 1 0 0 0 0
4 2 1 0 0 0 0
5 4 2 0 0 0 0
6 4 1 0 0 0 0
M END
But if you convert L-Alanin-2D.sdf to SDF/Mol, you will see that the wedge bond is changed to bond #2 connecting the stereo center to atom "C".
herong$ obabel L-Alanin-2D.sdf -o sdf
L-Alanin
OpenBabel02052108272D
2D Stereoinformation: Stereo Code
6 5 0 0 1 0 0 0 0 0999 V2000
3.6373 2.1000 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
2.4249 1.4000 0.0000 C 0 0 1 0 0 0 0 0 0 0 0 0
2.4249 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2124 2.1000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2124 3.5000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 1.4000 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2 1 1 0 0 0 0
2 3 1 1 0 0 0
4 2 1 0 0 0 0
5 4 2 0 0 0 0
6 4 1 0 0 0 0
M END
$$$$
1 molecule converted
According to Open Babel, the input uses an incorrect way to identify the stereo center. And it gives the correct way to identify stereo center.
If we load the same input data, L-Alanin-2D.sdf, into JSME (JavaScript Molecule Editor) at https://jsme-editor.github.io/dist/JSME_test.html, the wedge bond is not changed.
If we load the same input data, L-Alanin-2D.sdf, into pccdb.org Molecule Viewer at http://pccdb.org/tools/mol_viewer, the wedge bond is changed to a hash bond connecting the stereo center to atom "C". Positions of "N" and "C" are flipped to maintain the stereo conformation.
Graphical representations generated with Open Babel, JSME, and pccdb.org are presented below:
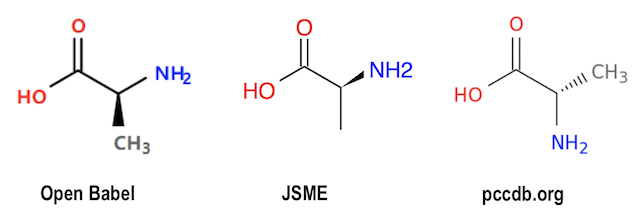
So the same SDF/Mol file with a stereo center specified with 2D Stereoinformation has 3 presentations. Who is more "correct"?
- Open Babel - Changed the wedge bond from "N" atom to "C".
- JSME - Kept the wedge bond at "N" as given in the input.
- pccdb.org - Flipped "N" and "C" positions and changed edge bond to hash bond on "C" atom.
Open Babel should provide an option to keep the 2D Stereoinformation as is from the input data, so we can control exactly how the molecule structure is presented in the graphical output.
Table of Contents
SMILES (Simplified Molecular-Input Line-Entry System)
Open Babel: The Open Source Chemistry Toolbox
Using Open Babel Command: "obabel"
Generating SVG Pictures with Open Babel
Substructure Search with Open Babel
Similarity Search with Open Babel
Fingerprint Index for Fastsearch with Open Babel
►Stereochemistry with Open Babel
Read Stereoinformation from Input with Open Babel
Stereo Perception Performed by Open Babel
Write Stereoinformation to Output by Open Babel
►Wedge-Hash Bond Changed by Open Babel
Hash Bond with Solid Line by Open Babel
Hash over Double Bond by Open Babel
Command Line Tools Provided by Open Babel
RDKit: Open-Source Cheminformatics Software
rdkit.Chem.rdchem - The Core Module
rdkit.Chem.rdmolfiles - Molecular File Module
rdkit.Chem.rdDepictor - Compute 2D Coordinates
rdkit.Chem.Draw - Handle Molecule Images
Molecule Substructure Search with RDKit
rdkit.Chem.rdmolops - Molecule Operations
Daylight Fingerprint Generator in RDKit
Morgan Fingerprint Generator in RDKit
RDKit Performance on Substructure Search
Introduction to Molecular Fingerprints
OCSR (Optical Chemical Structure Recognition)
AlphaFold - Protein Structure Prediction